無菌試験市場の規模・シェア分析 – 成長動向と予測 (2025年~2030年)
無菌性試験市場は、製品タイプ(機器、キット・試薬など)、試験タイプ(メンブレンろ過、直接接種など)、用途(医薬品・バイオ医薬品製造など)、実施形態(自社内試験、外部委託試験)、および地域(北米、欧州、アジア太平洋など)によってセグメント化されています。市場規模と予測は、金額(米ドル)で提供されます。

※本ページの内容は、英文レポートの概要および目次を日本語に自動翻訳したものです。最終レポートの内容と異なる場合があります。英文レポートの詳細および購入方法につきましては、お問い合わせください。
*** 本調査レポートに関するお問い合わせ ***
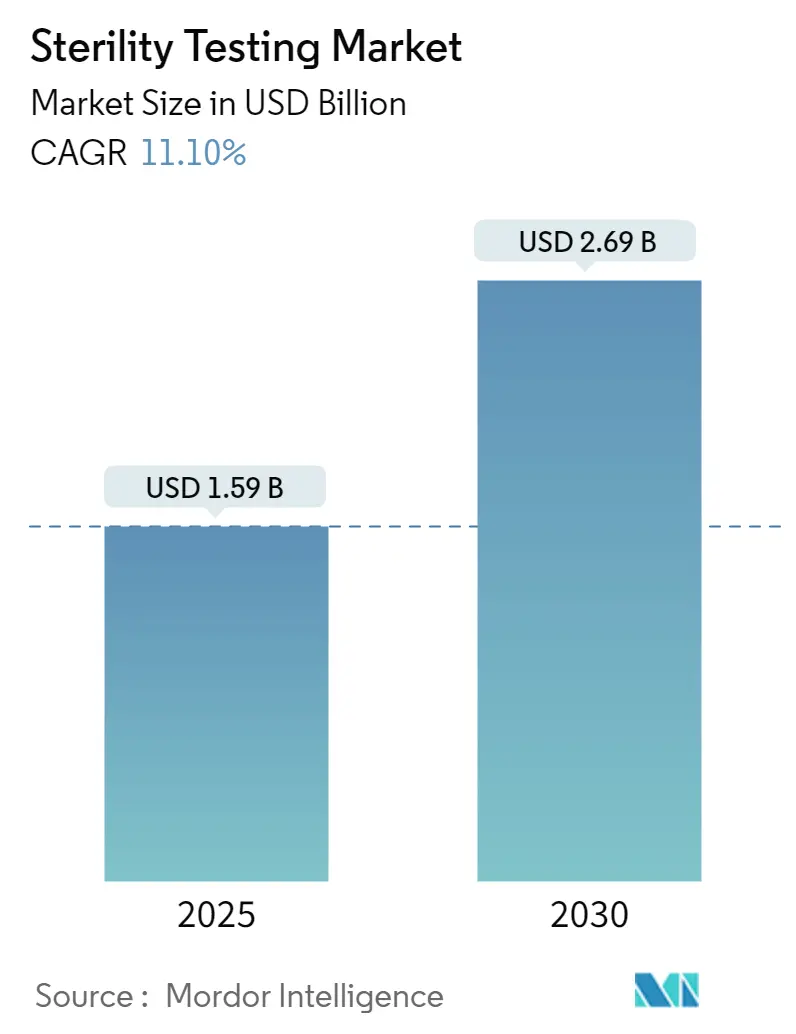
無菌性試験市場は、2025年に15.9億米ドルと評価され、2030年には26.9億米ドルに達すると予測されており、予測期間中の年平均成長率(CAGR)は11.1%で成長すると見込まれています。この市場は、製品タイプ(機器、キット・試薬、サービスなど)、試験タイプ(メンブレンろ過、直接接種、迅速無菌試験など)、用途(医薬品・バイオ医薬品製造、アウトソーシングCDMO試験など)、実施形態(社内試験、アウトソーシング/受託試験)、および地域(北米、欧州、アジア太平洋など)によってセグメント化されています。北米が最大の市場であり、アジア太平洋地域が最も急速に成長する市場です。市場の集中度は中程度です。
市場概要と主要動向
この市場の成長は、EU GMP Annex 1におけるゼロCFU(コロニー形成単位)要件の厳格化、複雑なバイオ医薬品パイプラインの商業化、および医薬品から患者へのサイクルを短縮する迅速リリース法の導入によって推進されています。細胞・遺伝子治療への継続的なベンチャーキャピタル投資、公的部門によるワクチン調達の増加、および無菌充填・仕上げ能力の受託パートナーへの移行も需要を活性化させています。メンブレンろ過は依然として確立された地位を維持していますが、迅速微生物検出プラットフォームは規制当局の支持を得ており、数週間かかっていたバッチ処分を数時間に短縮しています。北米の洗練された規制エコシステムが市場を牽引する一方で、アジア太平洋地域の新興メガプラント、優遇税制、および調和された薬局方更新が、同地域を最高のCAGRへと押し上げています。
市場を牽引する要因(Drivers):
* 高度なバイオ医薬品パイプラインに対する厳格なGMP要件の強化(CAGRへの影響度:+2.80%): EU GMP Annex 1の改訂により、ガイドラインが大幅に拡張され、グレードA環境におけるゼロCFU許容が正式化されました。これにより、アイソレーター、生菌モニタリング、滅菌前後の完全性試験(PUPSIT)への設備投資が急増しています。EMAとFDA間の調和は、多国籍メーカーがバリデーションマスタープランを標準化し、ロットリリース決定を加速させることを可能にしています。
* 迅速リリース試験を必要とする細胞・遺伝子治療の商業バッチの急増(CAGRへの影響度:+2.10%): 米国では1,200以上の臨床試験が進行中であり、自家細胞治療の承認が相次いでいることから、生細胞の効力を保護するために4時間以内の無菌性確認が求められています。bioMérieuxのSCANRDIは、固相サイトメトリーを利用して、培養不可能な単一の生菌を検出でき、結果までの時間を14日から150分未満に短縮し、USP <1223>の受容基準を満たしています。
* 社内QCからアウトソーシングCDMO無菌性サービスへの移行(CAGRへの影響度:+1.90%): 医薬品業界は、大規模な無菌性分析のために専門パートナーへの依存を強めています。Eurofinsは現在、45以上のGMPラボを運営し、無菌性、エンドトキシン、粒子試験を統合したワンストップハブを提供しています。このモデルは、熟練労働者不足から医薬品所有者を保護し、大陸をまたいだ24時間体制の冗長性を確保しています。
* 偽陽性を抑制するモジュラーアイソレーターシステムの採用(CAGRへの影響度:+1.50%): モジュラーアイソレーターはISO 14644-7に準拠し、グレードAの一方向気流を維持し、以前は偽陽性調査の35%を占めていたオペレーター起因の逸脱を削減します。IonHP+除染サイクルは、塩素残留物なしで15分以内に6-log以上の微生物減少を達成します。
* 迅速微生物学的手法に対する規制当局の推進(CAGRへの影響度:+1.30%): EU GMP Annex 1を触媒として、世界的に迅速微生物学的手法の検証が推進されています。
* シングルユース技術対応試験キットの需要増加(CAGRへの影響度:+1.00%): 世界中のバイオ医薬品ハブで需要が高まっています。
市場の成長を阻害する要因(Restraints):
* クラスBアイソレーターインフラの高い設備投資コスト(CAGRへの影響度:-1.80%): 自動リークテストモジュールを備えたデュアルチャンバーアイソレーターの取得には、バリデーションや年間サービス契約を除いて30万米ドル以上の費用がかかります。
* 公定試験基準の世界的な調和の限定性(CAGRへの影響度:-1.20%): USP、EP、JP、中国薬局方間の相違は、重複したバリデーション、消耗品在庫の増加、多国籍企業が非同期的な改訂にさらされる原因となっています。
* 新興市場における熟練した微生物学者の深刻な不足(CAGRへの影響度:-1.00%): アジア太平洋、ラテンアメリカ、中東・アフリカ地域で顕著です。
* 直接接種試験における偽陽性リスクによるリリース遅延(CAGRへの影響度:-0.80%): 大量生産地域でより大きな影響があります。
セグメント分析
* 製品タイプ別:
* サービスは10.8%のCAGRで拡大しており、微生物学者の不足の中で高度なアッセイをアウトソーシングしたいという業界の意欲を反映しています。主要なCDMOがQCスイートを無菌充填ラインの隣に組み込むことで、「製造場所での試験」パラダイムが可能になり、物流の滞留が削減されています。
* キット・試薬は2024年に50.7%の収益シェアを占め、中小規模メーカーの分散型品質管理ポイントに対応することで堅調を維持しています。シングルユースマニホールド、変色培養培地、既製の0.45 µm親水性PVDFメンブレンなどが、自動化が進む中でもその関連性を保っています。
* 機器は最も収益シェアが小さいものの、最も高いイノベーション指数を示しています。Growth Directモジュールは、126カセットを同時に培養し、AI画像分析を統合することで、高い信頼性閾値で48時間以内に公式の無菌性読み取りを提供します。
* 試験タイプ別:
* メンブレンろ過は2024年に71.4%の使用率を維持しており、低粘度の注射剤や眼科用製品に好まれています。その確立された薬局方での位置づけ、シンプルな消耗品、0.45 µmの公称孔径は、一貫したバリデーションを容易にします。しかし、14日間の培養期間はバイオ医薬品の貯蔵寿命と競合します。
* そのため、無菌性試験市場では、USP <71>の代替方法の文言に基づいて検証された迅速な無菌性試験方法が、バイオ医薬品の貯蔵寿命の課題に対応する解決策として注目を集めています。
本レポートは、医薬品、バイオ医薬品、医療機器の最終バッチにおける微生物の不在を確認するための無菌性試験市場に焦点を当てています。この市場は、キット、試薬、機器、および外部委託されたラボサービスから生じる収益で構成され、17カ国を対象に米ドルで評価されています。ただし、環境モニタリング消耗品や上流生産中に実施される工程内バイオバーデン試験は対象外とされています。
市場規模は2025年に15.9億米ドルと評価され、2030年までに26.9億米ドルに達すると予測されています。特に、外部委託サービスが最も急速に成長しており、製薬企業が専門知識を求めてCDMO(医薬品受託製造開発機関)に移行する中、2030年まで年平均成長率(CAGR)10.8%で進展すると見込まれています。
市場の成長を牽引する主な要因としては、以下の点が挙げられます。
* 先進的なバイオ医薬品パイプラインに対する厳格なGMP(医薬品製造管理および品質管理基準)要件の強化。
* 細胞・遺伝子治療薬の商業バッチにおける、迅速な製品リリースを可能にする無菌性試験の需要急増。
* 社内品質管理からCDMOへの外部委託への移行。
* 偽陽性率を低減するモジュラー式アイソレーターシステムの採用。
* 規制当局による検証済み迅速微生物試験法の推進。
* シングルユース技術に対応した試験キットへの需要増加。
一方で、市場の成長を抑制する要因には、以下の課題があります。
* クラスBアイソレーターインフラの高額な初期費用。
* 公定試験基準の国際的な調和の限定性。
* 新興市場における熟練した微生物学者の深刻な不足。
* 直接接種試験における偽陽性リスクによる製品リリースの遅延。
迅速無菌性試験は、保存期間が限られている細胞・遺伝子治療薬の製品リリースを加速させるため、その採用が拡大しています。また、EU GMP Annex 1の「ゼロCFU」要件は、製造業者にアイソレーターのアップグレード、PUPSIT(事前使用時完全性試験)の導入、高度な環境モニタリングシステムの設置を促し、新たな機器やサービス契約の需要を生み出しています。偽陽性結果を低減するための主要技術としては、モジュラー式アイソレーターシステム、AI搭載コロニーカウンター、固相サイトメトリープラットフォームが挙げられ、これらは手作業の介入と汚染リスクを減らし、関連コストを抑制します。
本レポートでは、市場を製品タイプ(機器、キット・試薬、サービス)、試験タイプ(メンブレンろ過、直接接種、迅速無菌性試験)、用途(医薬品・バイオ医薬品製造、医療機器製造など)、実施形態(社内試験、外部委託試験)、および地域(北米、欧州、アジア太平洋、中東・アフリカ、南米)別に詳細に分析しています。地域別では、北米が市場収益の42.3%を占め最大のシェアを保持しており、アジア太平洋地域が2030年まで年平均成長率9.7%で最も急速に成長する地域と予測されています。
競争環境については、市場集中度、市場シェア分析、およびbioMerieux SA、Charles River Laboratories、Merck KGaA、Sartorius AG、SGS SA、Thermo Fisher Scientificなどを含む主要企業18社のプロファイル(事業概要、財務状況、製品・戦略、最近の動向など)が含まれています。
調査は、医薬品製造工場や地域契約ラボの品質管理者へのインタビューを含む一次調査と、公定書、貿易データ、企業報告書などの二次調査を組み合わせて実施されています。市場規模の算出と予測は、グローバルな注射剤バッチ量と平均試験回数を需要プールに変換し、平均販売価格で評価するトップダウンモデルと、サプライヤーの集計やチャネルチェックによるボトムアップ検証を組み合わせて行われています。データは厳格な検証プロセスを経て、毎年更新されることで、信頼性の高い情報を提供しています。

1. はじめに
- 1.1 調査の前提と市場の定義
- 1.2 調査範囲
2. 調査方法
3. エグゼクティブサマリー
4. 市場概況
- 4.1 市場概要
- 4.2 市場の推進要因
- 4.2.1 高度なバイオ医薬品パイプラインに対する厳格なGMPアップグレード
- 4.2.2 迅速なリリース滅菌試験を必要とする細胞・遺伝子治療の商業バッチの急増
- 4.2.3 社内QCから外部委託CDMO滅菌サービスへの移行
- 4.2.4 偽陽性率を低減するモジュラーアイソレーターシステムの採用
- 4.2.5 検証済み迅速微生物学的検査法に対する規制当局の推進
- 4.2.6 シングルユース技術対応の検査キットに対する需要の増加
- 4.3 市場の阻害要因
- 4.3.1 クラスBアイソレーターインフラの高額な設備投資コスト
- 4.3.2 薬局方試験基準の国際的な調和の限定性
- 4.3.3 新興市場における熟練した微生物学者の深刻な不足
- 4.3.4 直接接種試験における偽陽性リスクによるリリース遅延
- 4.4 サプライチェーン分析
- 4.5 規制環境
- 4.6 技術的展望
- 4.7 ポーターの5つの力分析
- 4.7.1 新規参入者の脅威
- 4.7.2 買い手の交渉力
- 4.7.3 供給者の交渉力
- 4.7.4 代替品の脅威
- 4.7.5 競争の激しさ
5. 市場規模と成長予測(金額)
- 5.1 製品タイプ別
- 5.1.1 機器
- 5.1.2 キット&試薬
- 5.1.3 サービス
- 5.2 テストタイプ別
- 5.2.1 メンブレンろ過
- 5.2.2 直接接種
- 5.2.3 迅速無菌性試験
- 5.3 用途別
- 5.3.1 医薬品・バイオ医薬品製造
- 5.3.2 医療機器製造
- 5.3.3 その他
- 5.4 モード別
- 5.4.1 社内試験
- 5.4.2 外部委託/受託試験
- 5.5 地域別
- 5.5.1 北米
- 5.5.1.1 米国
- 5.5.1.2 カナダ
- 5.5.1.3 メキシコ
- 5.5.2 欧州
- 5.5.2.1 ドイツ
- 5.5.2.2 英国
- 5.5.2.3 フランス
- 5.5.2.4 イタリア
- 5.5.2.5 スペイン
- 5.5.2.6 その他の欧州
- 5.5.3 アジア太平洋
- 5.5.3.1 中国
- 5.5.3.2 日本
- 5.5.3.3 インド
- 5.5.3.4 韓国
- 5.5.3.5 オーストラリア
- 5.5.3.6 その他のアジア太平洋
- 5.5.4 中東およびアフリカ
- 5.5.4.1 GCC
- 5.5.4.2 南アフリカ
- 5.5.4.3 その他の中東およびアフリカ
- 5.5.5 南米
- 5.5.5.1 ブラジル
- 5.5.5.2 アルゼンチン
- 5.5.5.3 その他の南米
- 5.5.5.3.1 サウジアラビア
6. 競合情勢
- 6.1 市場集中度
- 6.2 市場シェア分析
- 6.3 企業プロファイル(グローバル概要、市場概要、主要セグメント、利用可能な財務情報、戦略情報、主要企業の市場ランキング/シェア、製品&サービス、および最近の動向を含む)
- 6.3.1 bioMerieux SA
- 6.3.2 Charles River Laboratories
- 6.3.3 Merck KGaA
- 6.3.4 Sartorius AG
- 6.3.5 SGS SA
- 6.3.6 Sotera Health (Nelson Laboratories)
- 6.3.7 STERIS Plc
- 6.3.8 Thermo Fisher Scientific
- 6.3.9 Laboratory Corporation of America Holdings
- 6.3.10 WuXi AppTec
- 6.3.11 Rapid Micro Biosystems Inc.
- 6.3.12 Pace Analytical
- 6.3.13 Eurofins Scientific
- 6.3.14 Pacific BioLabs
- 6.3.15 Lonza Group
- 6.3.16 Boston Analytical
- 6.3.17 Bio-Outsource (SGS)
- 6.3.18 Toxikon Europe
7. 市場機会と将来展望
*** 本調査レポートに関するお問い合わせ ***

無菌試験は、医薬品、医療機器、再生医療等製品など、無菌性が求められる製品において、その製品中に生菌微生物が存在しないことを科学的に証明するための極めて重要な品質管理試験です。この試験の目的は、製品が患者に投与または使用された際に、微生物による感染症のリスクを排除し、患者の安全を確保することにあります。無菌試験は、製品の最終品質を保証する上で不可欠な工程であり、各国の薬局方(日本薬局方、米国薬局方、欧州薬局方など)によって厳格な実施方法が規定されています。
無菌試験には主に二つの方法があります。一つは「直接接種法」で、試験対象の製品を直接、微生物の増殖に適した培地に接種し、一定期間培養することで微生物の有無を確認する方法です。この方法は、製品の量が比較的少なく、培地の増殖を阻害する成分が含まれていない場合に適しています。もう一つは「メンブレンフィルター法」で、試験対象の製品をメンブレンフィルターに通して微生物を捕捉し、フィルターを洗浄して製品由来の阻害物質を除去した後、フィルターを培地に接触させて培養する方法です。この方法は、製品の量が多い場合や、抗菌性物質など培地の増殖を阻害する成分が含まれている場合に特に有効です。使用される培地としては、好気性菌および真菌の検出に適した大豆カゼインダイジェスト培地(SCD培地)と、嫌気性菌を含む幅広い微生物の検出に適した流動チオグリコール酸培地(FTM培地)が一般的です。これらの培地は、それぞれ異なる温度条件下で規定された期間(通常14日間)培養され、微生物の増殖が観察されないことをもって「無菌」と判断されます。試験の実施に先立ち、試験法が製品の微生物検出を阻害しないことを確認するための「試験法適合性試験(培地適合性試験)」が必須となります。
無菌試験の用途は多岐にわたります。最も主要な用途は、注射剤、点眼剤、輸液剤、ワクチンなどの無菌医薬品の最終製品検査です。これらの製品は、直接体内に投与されるため、無菌性が絶対条件となります。また、手術用具、カテーテル、インプラントなどの医療機器も、体内に挿入されるため無菌試験の対象となります。近年では、細胞治療製品や遺伝子治療製品といった再生医療等製品においても、その特性に応じた無菌試験が実施され、患者への安全性が確保されています。さらに、医薬品製造工程で使用される原材料や中間製品、製造環境のモニタリングサンプルなどに対しても、工程管理の一環として無菌性評価が行われることがあります。
無菌試験に関連する技術は、その信頼性と効率性を高めるために進化を続けています。まず、製品が無菌状態を保つための「無菌操作法」や、製造環境の清浄度を維持・管理する「環境モニタリング」は、無菌試験の結果を左右する重要な要素です。クリーンルームの設計、HEPAフィルターの性能管理、作業員の教育訓練などがこれに含まれます。また、製品を無菌化するための「滅菌法」(高圧蒸気滅菌、放射線滅菌、エチレンオキサイドガス滅菌、乾熱滅菌など)も、無菌試験と密接に関連しています。近年注目されているのは「迅速微生物試験法(RMM)」です。これは、従来の培養法に比べて短時間で微生物の有無や種類を検出する技術で、ATPルミネッセンス法、フローサイトメトリー法、PCR法などが開発されています。これらの迅速法は、製品の早期出荷や製造工程のリアルタイム管理に貢献しますが、最終製品の無菌試験においては、そのバリデーションと規制当局の承認が課題となります。さらに、製品の無菌状態を維持するための「容器完全性試験(CCIT)」も、無菌試験と並行して実施される重要な試験です。
市場背景としては、世界の医薬品市場、特にバイオ医薬品や再生医療等製品の成長に伴い、無菌試験の需要は拡大の一途を辿っています。これらの製品は、多くが無菌製剤であり、その品質保証には厳格な無菌試験が不可欠です。また、グローバルな医薬品サプライチェーンの複雑化により、各国・地域の規制要件への適合がより一層求められています。無菌試験は、製品の品質保証における最終防衛線であり、その結果が製品の市場投入を左右するため、製薬企業にとっては非常にコストと時間のかかる工程でもあります。試験の失敗は、製品のリコールや製造停止、企業の信頼失墜に直結するため、その重要性は計り知れません。規制当局は、無菌試験の実施方法、バリデーション、結果の解釈について詳細なガイドラインを提示しており、企業はこれらを遵守する必要があります。
将来の展望として、無菌試験はさらなる自動化と迅速化の方向へと進化していくと考えられます。人為的ミスを排除し、試験の再現性と信頼性を向上させるためのロボット技術や自動化システムの導入が進むでしょう。これにより、試験の効率化とコスト削減が期待されます。また、前述の迅速微生物試験法は、その適用範囲を広げ、最終製品の無菌試験においても、従来の培養法に代わる、あるいは補完する技術として確立される可能性があります。そのためには、これらの新しい試験法のバリデーションデータの蓄積と、規制当局による承認が不可欠です。さらに、プロセスアナリティカルテクノロジー(PAT)やクオリティバイデザイン(QbD)の概念が浸透するにつれて、製造工程全体で無菌性を確保し、最終製品の無菌試験への依存度を低減するアプローチも強化されるでしょう。これは、最終試験で無菌性を確認するだけでなく、製造プロセスそのものに無菌性を組み込むという考え方です。AIやビッグデータ解析の活用により、試験データの解析やリスク評価の精度が向上し、より効率的で信頼性の高い無菌保証システムが構築されることも期待されます。これらの技術革新は、患者へのより安全な製品提供と、製薬産業の持続的な発展に貢献していくことでしょう。